

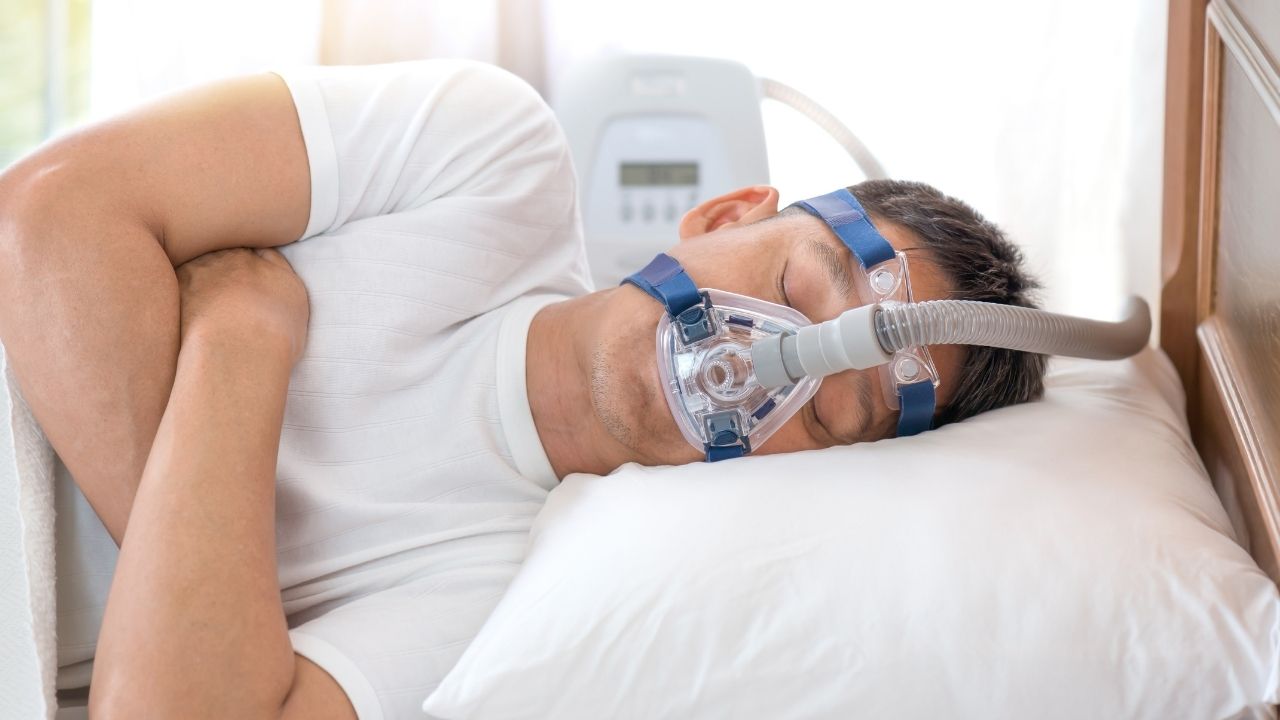
 Philips Respironics (Philips) is a major manufacturer of the CPAP and BiPAP machines that help people with central sleep apnea and obstructive sleep apnea. Philips also makes ventilators that have no doubt saved lives in hospitals and other healthcare settings. These machines use a special type of foam to reduce the sound and vibration coming from these devices. Unfortunately, the foam Philips chose years ago can degrade and injure users, which eventually led to a 2021 CPAP recall. Let’s take a look at a recent development concerning Philips and its ventilator, CPAP and BiPAP products.
Philips Respironics (Philips) is a major manufacturer of the CPAP and BiPAP machines that help people with central sleep apnea and obstructive sleep apnea. Philips also makes ventilators that have no doubt saved lives in hospitals and other healthcare settings. These machines use a special type of foam to reduce the sound and vibration coming from these devices. Unfortunately, the foam Philips chose years ago can degrade and injure users, which eventually led to a 2021 CPAP recall. Let’s take a look at a recent development concerning Philips and its ventilator, CPAP and BiPAP products.
A Brief Overview of How We Got Here
In June 2021, the U.S. Food and Drug Administration (FDA) issued a Safety Communication notifying the public that Philips had recalled certain BiPAP, CPAP and ventilator machines. There were concerns that the polyester-based polyurethane (PE-PUR) foam used in these devices could break down, allowing small bits of foam to enter a person’s body. The FDA also mentioned that the foam could off-gas and release harmful gasses into the machine’s airflow causing additional harm.
In response to the CPAP recall, the FDA asked Philips to do several things, including:
- Let the FDA inspect the Philips manufacturing facilities.
- Take additional steps to notify health professionals who use or prescribe the affected products.
- Have an independent laboratory conduct additional testing concerning the health risks presented by the PE-PUR foam.
Philips agreed to these requests and recently reported its findings from its independent laboratory testing to the FDA.
Philips Testing and the FDA’s Response
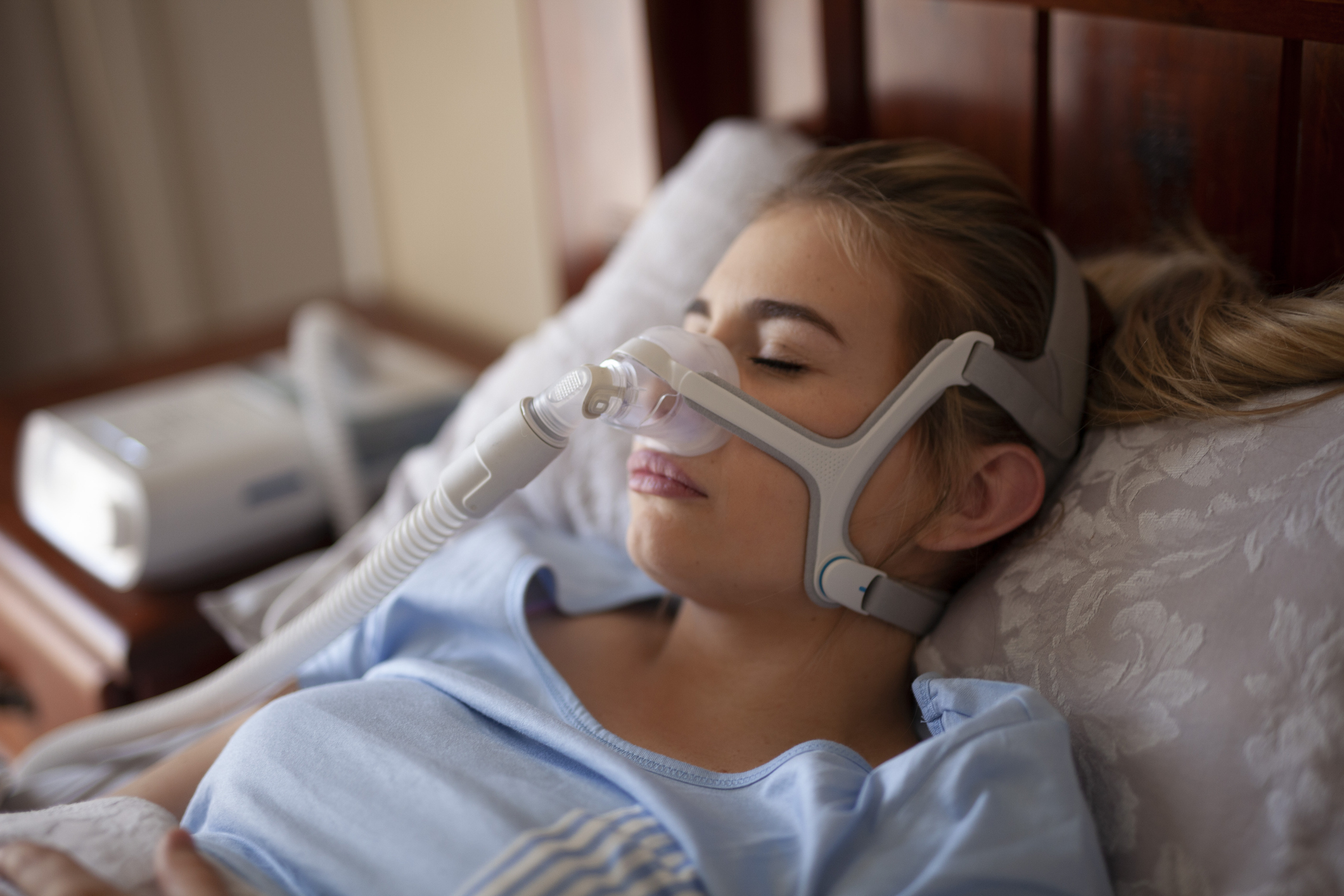 Throughout 2021, 2022 and 2023, Philips provided regular updates concerning the testing being done by its independent laboratories. This testing examined two types of PE-PUR foam used in the recalled devices and assessed the risk they presented when they broke down and off-gassed VOCs (volatile organic compounds) or introduced particulates (tiny bits of foam material) into the airflow.
Throughout 2021, 2022 and 2023, Philips provided regular updates concerning the testing being done by its independent laboratories. This testing examined two types of PE-PUR foam used in the recalled devices and assessed the risk they presented when they broke down and off-gassed VOCs (volatile organic compounds) or introduced particulates (tiny bits of foam material) into the airflow.
Philips used five independent laboratories across the United States and Europe and the testing examined both new and used devices, as well as devices cleaned with ozone, which the FDA suspects might make the PE-PUR foam problem worse.
Also tested were “lab-aged” machines. These are machines that were exposed to temperatures (90 degrees Celsius) and relative humidity (95%) that are far higher than what’s typically found in people’s bedrooms. If you’re not familiar with the metric system, 90 degrees Celsius is more than 190 degrees Fahrenheit.
When testing Type A foam, the laboratories concluded that any particulate or VOC exposure was “unlikely to result in an appreciable harm to health in patients.” When testing Type B foam (which accounted for 5% of the recalled machines), the laboratories concluded that “no overall risk conclusion [were] available at this time.”
As you might expect, the FDA was not satisfied with these results and asked that Philips complete more testing. Philips has agreed to this and while the FDA awaits additional test data, the FDA stands by its original patient recommendations concerning the use of the recalled machines.
If you’re curious about the nitty-gritty details of this testing, you can review the results in Philips’ July 2023 report. Just be warned that the content is a bit dense, although reading this report would work as a great sleep aid if you’re having trouble falling asleep.
I’ll provide more updates as they arrive, including major news concerning the CPAP recall or the ongoing litigation. If you have any questions on what your legal options might be if you’ve used a recalled machine, don’t hesitate to reach out by calling me. My direct line in the office is 919.334.6277.